

In recent years, the regulatory landscape for aesthetic device manufacturers has evolved significantly. Devices previously categorized as wellness products, including many energy-based platforms, are now subject to medical-grade scrutiny worldwide.
Consumer demand for non-invasive treatments is surging. The global market for aesthetic medical devices is expected to surpass $19.42 billion by 2035, expanding at a CAGR of 3.2%. However, behind the commercial opportunity lies growing regulatory risk. As these devices deliver energy deeper into tissue, their physiological effects and clinical claims are drawing heightened regulatory oversight across global markets, including the EU, U.S., Japan, Canada, Brazil, Australia, and key ASEAN, GCC, and LATAM markets.
From the FDA’s 125% rise in device recalls over the past decade to the EU’s Annex XVI extension of CE Marking to non-medical technologies, the message is clear: regulatory missteps now carry real commercial risk. For medical aesthetic device manufacturers, preemptive compliance is essential to minimize time to market and fully leverage their global scale-up potential.
This blog serves as a technical roadmap for QARA leaders and MedTech founders navigating this evolving landscape of aesthetic device regulation.
Regulatory Triggers for Energy-Based Aesthetic Devices
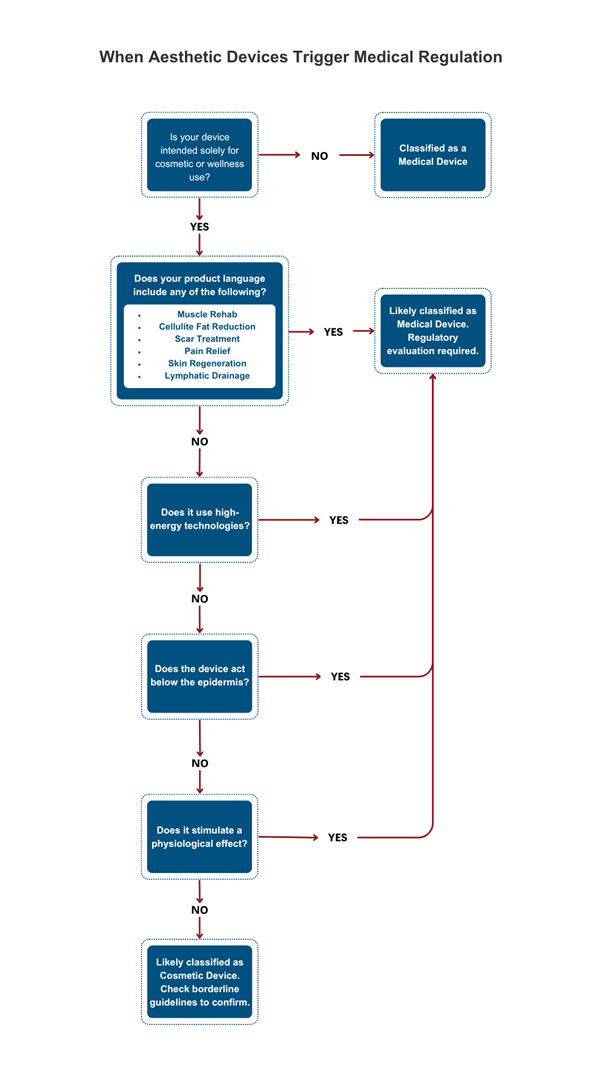
Energy-based aesthetic devices deliver mechanical, thermal, or electrical energy to biological tissue to achieve defined cosmetic or therapeutic outcomes. However, when those effects cross into physiological impact zones, such as muscle contraction, fat disruption, or skin regeneration, they trigger medical device regulations.
Core Modalities Behind Modern Energy-Based Aesthetic Devices:
- Radiofrequency (RF): Delivers controlled thermal energy to stimulate collagen remodeling and dermal tightening
- Laser & IPL (Intense Pulsed Light): Uses focused light energy to ablate, coagulate, or photothermally treat skin tissues
- Ultrasound (including cavitation, HIFU): Provides mechanical vibrations or focused thermal effects for lipolysis or lifting
- Electrostimulation (EMS, TENS, IFT): Triggers muscle contraction or nerve stimulation for toning or pain relief
- Microcurrent therapy: Applies low-level electrical currents to enhance cellular repair and ATP production
Devices crossing from cosmetic action into dermal, adipose, or neuromuscular impact become candidates for stricter regulation.
Key Regulatory Triggers That Classify Aesthetic Devices as Medical
1. Intended Use & Claims
- The first and most critical classification driver is the stated intended use. Terms like “scar reduction,” “muscle rehabilitation,” “cellulite reduction,” or “fat breakdown” signal therapeutic or physiological intent and shift the device under formal medical regulation.
- Even subtle language, “improves tone,” “revitalizes skin,” or “tightens fascia”, can raise flags if the claimed mechanism implies action below the epidermis.
- Claims must also be mapped against mechanism of action. For instance, a device claiming “lymphatic drainage” but using EMS pulses to stimulate muscle contraction may still be viewed as therapeutic.
- Across regions, regulators consider not just claims made but inferred intent from user manuals, UI, and third-party marketing.
2. Mode of Action & Tissue Penetration
- Classification increases as the device’s action moves deeper into the tissue, beyond superficial heating or micro-massage to affecting dermal, muscular, or adipose layers.
- For example, HIFU or RF systems targeting SMAS (superficial muscular aponeurotic system) or subcutaneous fat generally fall under Class IIb in the EU and Class II (or De Novo) in the U.S
- Ablative or semi-ablative devices (e.g., fractional lasers) also trigger more stringent risk of categorization due to their penetration and disruption of skin barriers.
3. Physiological Impact
- Devices that generate meaningful biological responses, such as thermal coagulation, neuromuscular stimulation, lipolysis, or tissue ablation, are inherently higher risk
- Regulators may demand evidence of both efficacy (via clinical studies or predicate devices) and safety (thermal maps, cytotoxicity, animal data)
- The regulatory gaze is widening, for example, in the EU under MDR, even non-medical devices demonstrating these impacts fall under Annex XVI requirements.
4. Risk Profile
- Risk Class assignments are driven by both the probability and severity of potential harm
- Higher risk classes (EU IIb, US Class II with special controls) demand additional elements like:
- a. Safety interlocks
- b. EMC/IEC compliance (e.g., IEC 60601-1-2)
- c. Labeling with contraindications, protective eyewear, pre-treatment testing
- Risk-benefit analysis must be robust and documented within the device’s Risk Management File (per ISO 14971:2019).
Regional Compliance Strategies for Energy-Based Aesthetic Devices
A. European Union – MDR & Annex XVI
Under EU MDR 2017/745, aesthetic devices without medical purpose, but with similar risk profiles, fall under Annex XVI. These include:
- RF body contouring and wrinkle reduction
- Laser/IPL hair or tattoo removal
- Ultrasonic lipolysis
Classification:
- Class IIa or IIb, depending on penetration and energy level
Requirements:
- CE Marking through Notified Body
- ISO 13485:2016-certified QMS
- Clinical Evaluation Report (CER)
- PMS and vigilance systems
Specific add-ons for Italy – a growing market within the EU:
- Registration with Ministero della Salute
- IFUs and labels must be in Italian
Emerging Trend: As of 26 May 2025, UDI labelling requirements for Class I devices under the EU MDR are fully in effect, making UDI compliance mandatory for all new products placed on the EU market.
B. United States – FDA (CDRH)
The USFDA classifies based on physiological effect, not product labeling. Aesthetic lasers, RF systems, or EMS stimulators are regulated under 21 CFR Part 878. Common Class II Devices include:
- RF skin tightening
- Laser resurfacing
- Ultrasound fat disruption
- EMS devices for muscle toning
Pathway:
- 510(k) premarket notification with predicate comparison
Requirements:
- 21 CFR 820-compliant QMS
- IEC 60601 family electrical safety standards
- Software validation (if applicable)
- UDI and MDR compliance
Note: While many aesthetic devices are cleared via the 510(k) pathway, higher-risk technologies, such as those with deeper dermal or neuromuscular effects, may require Premarket Approval (PMA), USFDA’s most rigorous pathway. Although less common in aesthetics, misclassified devices may be recalled if post-market review suggests a PMA-level risk profile.
C. South Korea – MFDS
The MFDS uses a 4-class system based on device risk.
Energy-based aesthetic devices typically fall under Class 2 or 3, requiring:
- Pre-market Certification or Approval
- K-GMP (Korea-GMP) certification
- Appointing a Korea License Holder (KLH)
- Local safety/EMC testing and Korean-language documentation
- Clinical data (if novel mechanism or claim)
Potential Challenge: Strict scrutiny of claims like “lifting” or “slimming” can escalate classification or delay approvals.
Aesthetic Devices: Market Opportunity vs. Regulatory Complexity
| Region | Market Opportunity | Regulatory Complexity | Market-Specific Insights |
| USA | High | High | Strong demand; requires FDA clearance via 510(k), De Novo, or PMA depending on intended use and energy sources (e.g., lasers, RF, ultrasound). FDA considers many aesthetic devices as medical if they affect structure/function or claim therapeutic outcomes. |
| EU | High | High | Under EU MDR and Annex XVI, even non-medical aesthetic devices (e.g., body contouring, lasers) now face medical-grade compliance. CE marking is essential. Clinical evaluation and PMS/PMCF are key. |
| South Korea | Medium | Medium-High | Home to major beauty tech innovation. Devices must be approved by MFDS. Requires local clinical data; very strict advertising and efficacy claims scrutiny. |
| India | Medium-High | Medium | Rapidly expanding consumer base and aesthetic industry. CDSCO governs devices; some aesthetic devices classified as medical. Regulatory clarity improving post-2017 Medical Device Rules but still evolving. |
| Brazil | Medium | High | Aroval can be time-consuming. Local clinical testing may be required. Cosmetic regulations are strict if therapeutic claims are made. Many electro-medical / active devices must obtain INMETRO conformity certification (often to IEC 60601 standards) as a pre-requisite or parallel step to ANVISA registration. ANATEL certification must generally be completed before or in parallel to ANVISA registration if the product uses RF communications. |
| UAE | Medium | Low | Accepts CE/FDA approvals. Devices are often registered through MOHAP with simplified pathways. High per capita spend on aesthetics; medical tourism hub. |
| Australia | Low-Medium | Medium | TGA oversees approvals; often leverages existing FDA/CE approvals. Local sponsors are required. The market is quality-conscious but small in volume. |
Emerging and Frontier Markets for Aesthetic Devices
| Region | Market Opportunity | Regulatory Complexity | Market-Specific Insights |
| Vietnam | Medium | Medium | Growing demand for aesthetics among young consumers. MOH registration required; regulatory framework modelled on ASEAN MDD. |
| Indonesia | Medium | Medium-High | Rapid market growth, but BPOM (NADFC) has detailed requirements. Requires local representation and approval. |
| Mexico | Medium | Medium | COFEPRIS governs medical devices. Leverages some foreign approvals. Increasing interest in energy-based aesthetic procedures. |
| Saudi Arabia | Medium | Medium | SFDA requires registration; increasingly moving toward independent evaluation but still recognizes FDA/CE for many devices. |
| Turkey | Medium | Medium | Harmonized with EU MDR but requires a local authorized representative. Strategic location for Eurasian market access. |
Standards-Based Design & Risk Management for Aesthetic Devices
A ‘standards-first’ design approach not only accelerates global approvals; it builds safer, more consistent aesthetic devices. Regulatory leaders expect evidence that your design team is proactively applying globally recognized benchmarks. For example, EN ISO 14971:2019 + A11:2021 is now the harmonized risk-management standard under the EU MDR/IVDR. Using this harmonized version supports presumption of conformity to MDR risk-management requirements, and Notified Bodies note that early, full adoption of EN ISO 14971:2019-compliant processes helps streamline CE-mark review.
Designing to internationally recognized standards reduce regulatory risk across markets:
| Standard | Focus |
| ISO 13485:2016 | QMS Backbone |
| ISO 14791:2019 | End-to-end risk management |
| IEC 60601-1/-2 | Electrical safety & EMC |
| IEC 60825-1 | Laser Safety |
| IEC 62366 | Usability/human factors |
| IEC 62304 | Software lifecycle (for smart system) |
| ISO 10993 | Biocompatibility for contact materials |
Best Practices:
- Build a design traceability matrix linking
Input→ Output → Risk → Verification → Validation Maintain living DHF and Risk Management File (RMF) - Use global predicate mapping to minimize duplication across markets
Evaluating Diverse Strategies for Aesthetic Device Compliance
While expanding globally, device makers often face costly regulatory setbacks. Strategic missteps translate into business risk:
| Approach | Pros | Cons |
| “Global By Design” – Early Compliance for all target markets |
– Holistic design meets the strictest requirements (EU MDR, FDA, etc.) from the start – Easier entry into multiple markets without redesign – Fosters a culture of quality and readiness – One comprehensive tech file can be adapted globally |
– Higher initial R&D and QARA costs – Longer development cycle due to broad testing (e.g. both CE and FDA requirements) – Risk of over-engineering if some markets are not entered – Requires diverse regulatory expertise on the team early |
|
“One Market First” – Staged expansion (e.g. FDA first, then others) |
– Quicker initial launch to generate revenue or clinical experience in one market – Lower upfront compliance burden focused on one regulator – Spreads cost over time – Allows iteration from real-world use before international scale-up |
– Subsequent market entries may require design changes or additional testing (if the initial design is not fully compliant elsewhere) – Potential duplication of effort (multiple submissions and studies) – Competitors in other regions may advance while you focus on one market |
Many leading firms use a hybrid strategy. They identify a primary market but keep an eye on other major markets’ requirements to avoid major redesigns.
For example, a MedTech firm might initially seek FDA clearance but design the labeling and technical documentation to also satisfy MDR essentials, so that a CE marking process can follow swiftly. The right approach depends on resource capacity, device novelty, and business goals, but it must be a conscious choice informed by regulatory insight.
From Compliance to Commercial Impact: Building Aesthetic Devices That Succeed Globally
In the arena of energy-based aesthetic devices, the regulatory strategy directly defines commercial outcomes. Compliance efforts might seem expensive when targeting multiple high-barrier markets, yet when embedded early in design and planning, these efforts translate into operational clarity and long-term advantage.
Strategic alignment with global standards, paired with thoughtful claim language, predicate mapping, and risk management, helps streamline approvals, reduce rework, and support safer, more effective aesthetic technologies. Regulatory discipline not only protects users and patients, but also drives speed, scale, and profitability.
Syrma Johari MedTech supports aesthetic device manufacturers by integrating regulatory strategy into every stage of product development, from concept to global launch. With deep expertise in global regulatory frameworks, our team helps ensure faster approvals, reduced redesign cycles, and market-ready compliance documentation. Whether scaling in the U.S., EU, Asia or other emerging markets, we enable companies to navigate complex aesthetic device regulations with confidence and steady momentum.