

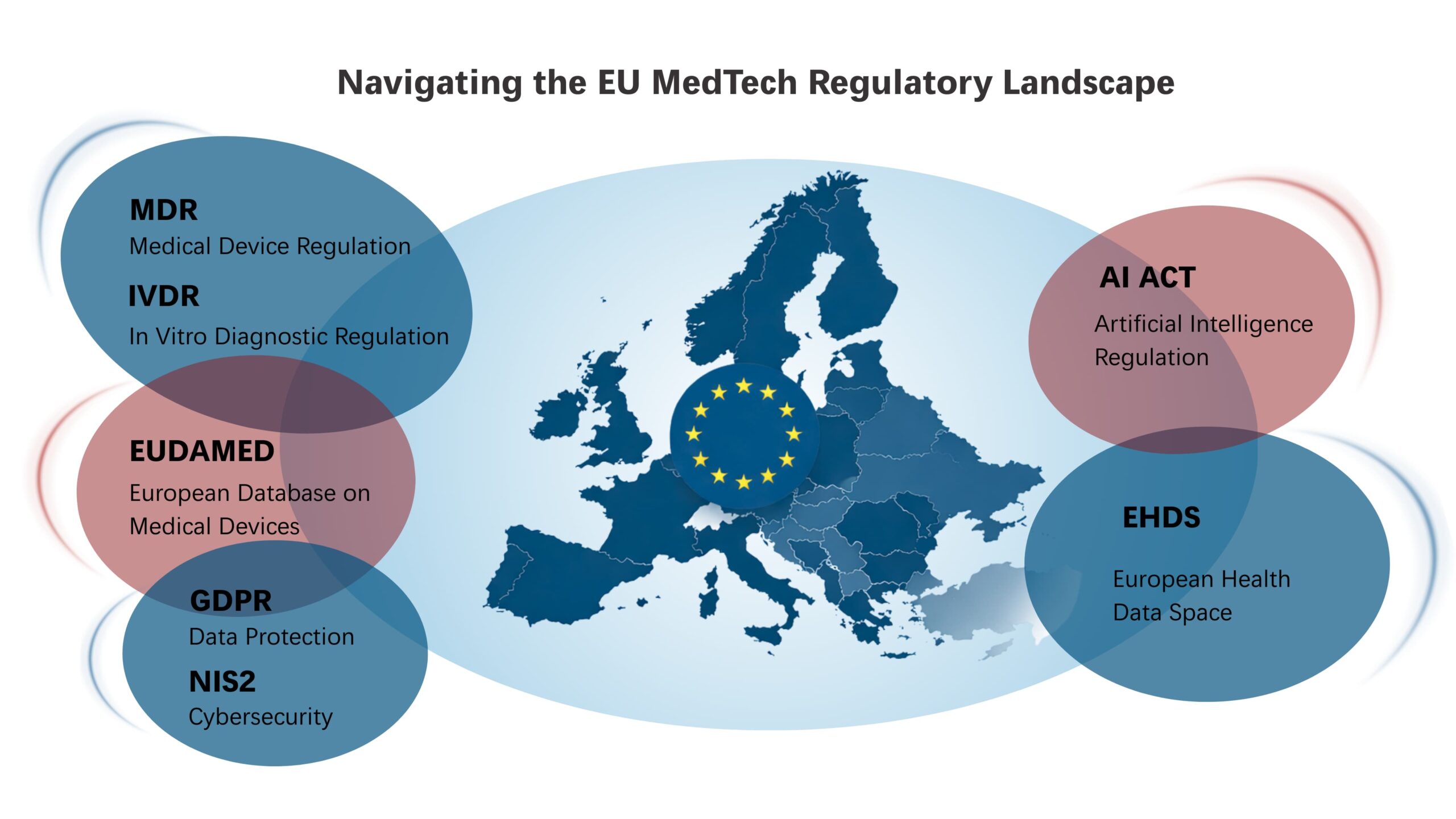
Europe is leading MedTech innovation across diverse segments, including AI-enabled imaging and diagnostics, advanced in vitro diagnostics, digital health and SaMD, medical robotics, and implantable technologies. Across these segments, new technologies are developed in alignment with regulatory frameworks such as the EU Medical Device Regulation (MDR), In Vitro Diagnostic Regulation (IVDR), the EU Artificial Intelligence Act (AI Act), European Database on Medical Devices (EUDAMED) and the European Health Data Space (EHDS). This ensures EU MedTech innovation advances alongside demonstrable safety, clinical performance and quality.
Roche Diagnostics exemplifies EU-first MedTech innovation in high-complexity in vitro diagnostics. As IVDR raised expectations around clinical performance, scientific validity and lifecycle evidence, Roche aligned assay design, clinical performance studies and manufacturing traceability for uninterrupted EU market access. Similarly, Siemens Healthineers launched AI-enabled imaging in Europe by designing software architectures that support controlled algorithm updates and traceability under MDR compliance. Clinical validation and post-market systems were structured to demonstrate and monitor real-world algorithm performance, while manufacturing quality systems governed hardware–software integration as products scaled. This approach enabled Siemens Healthineers to deploy AI technologies in the EU with consistent compliance as AI systems evolved.
Across these examples, sustained EU market success is closely linked to the integration of quality and regulatory strategy across the product lifecycle. For novel MedTech products operating without established precedent, this integration is the most challenging.
Regulatory Complexity Surrounding Next-Gen EU MedTech Innovation
Devices such as the Medtronic Micra leadless pacemaker, Edwards SAPIEN 3 transcatheter valve, and AI-based imaging platforms from Viz.ai and HeartFlow show that first-of-kind technologies can seamlessly enter the European market when innovators identify the unique challenges of EU regulatory frameworks and plan for them early. Breakthrough medical devices rarely align fully with legacy regulatory classifications. Novel mechanisms of action increase scrutiny across clinical evidence depth, risk–benefit justification and post-market surveillance obligations. Pioneering products also operate without predicate devices, established standards or clear Notified Body precedent.
In response to this technological uncertainty, EU regulators apply heightened expectations, including expanded clinical evaluation, increased emphasis on lifecycle evidence and stricter notified body requirements. For innovation-led MedTech companies in the EU, addressing this complexity requires an execution model that integrates design, manufacturing, and regulatory systems from the outset. This requirement can be structured as a six-layer innovation stack, in which all functions operate as one coordinated execution system.
The EU MedTech Innovation Stack: Design, Manufacturing and Regulatory Systems as a Single Execution System
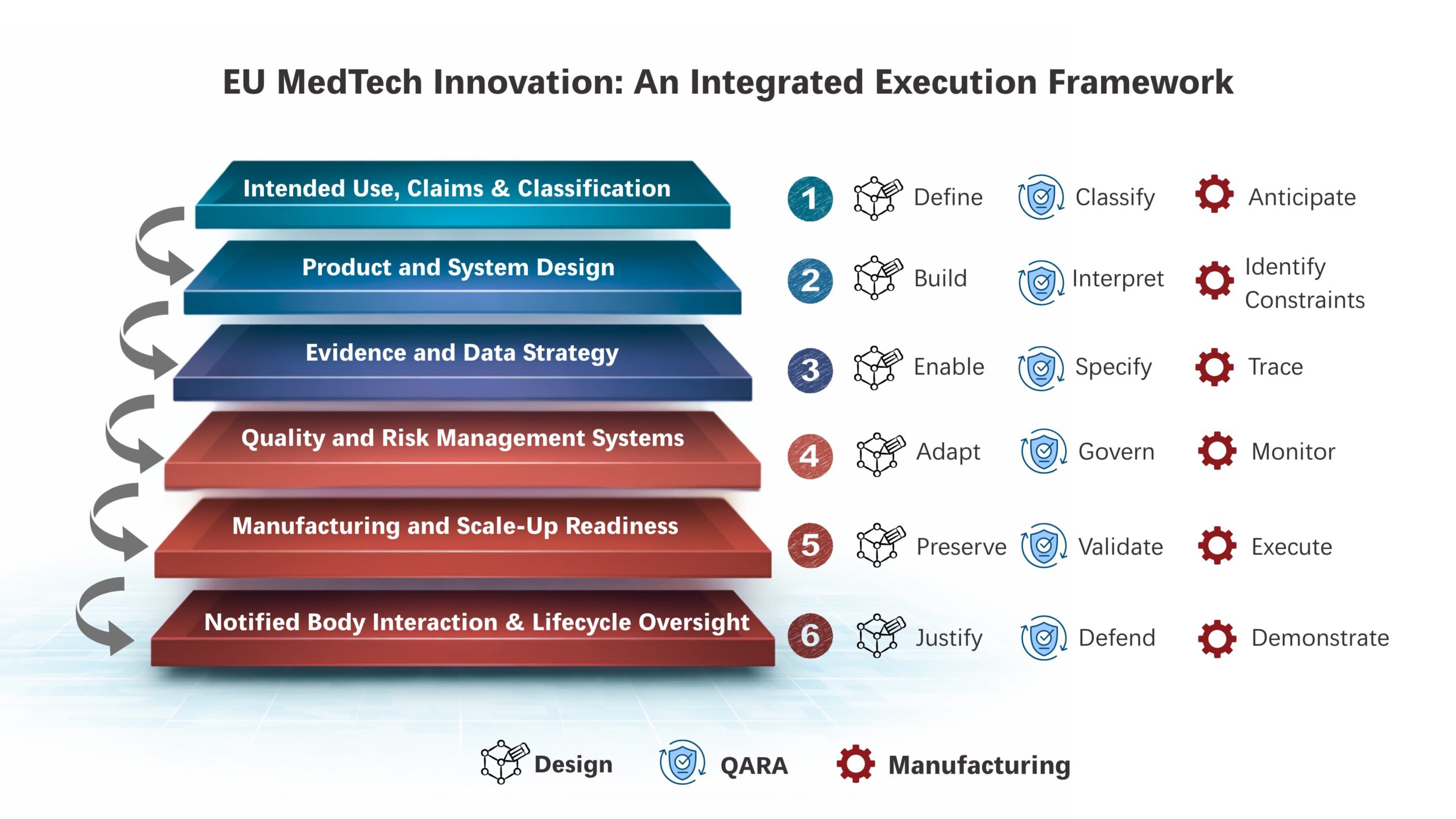
Layer 1: Intended Use, Claims and Classification Architecture
For groundbreaking EU MedTech innovation, intended use definitions and clinical claims establish the regulatory classification and set the boundary conditions for product design, evidence scope and manufacturing controls. Design teams must define functionality, clinical scope and performance limits at an early stage, while regulatory teams must map these decisions to MDR or IVDR classification rules, conformity assessment routes, and EUDAMED registration and transparency obligations. Manufacturing intent should develop in parallel, based on anticipated scrutiny and control requirements. Alignment at this layer establishes regulatory feasibility and execution stability across the lifecycle.
Layer 2: Product and System Design
For next-gen MedTech innovations, product architecture must evolve alongside regulatory interpretation. Under EU MDR/IVDR and emerging AI and digital health regulations, system architecture and software design choices should directly guide verification, validation, clinical evaluation and change management. Quality and regulatory teams must work alongside engineering to ensure design controls support traceability from intended use through risk management, verification, clinical performance and post-market outcomes. Early convergence at this layer prevents redesign and revalidation cycles and preserves change velocity under EU medical device regulations.
Layer 3: Evidence and Data Strategy
As per MDR and IVDR compliance, clinical evaluation, Post-Market Clinical Follow-Up (PMCF), and real-world data collection must operate as a continuous evidence system across the product lifecycle. Therefore, product design should enable appropriate data capture and performance monitoring capability. Meanwhile, quality and regulatory systems must define evidence expectations covering safety, clinical performance, and benefit–risk profile. Together, these functions should define Annex XIV–aligned clinical evaluation and PMCF plans and establish data pipelines for lifecycle risk management. As the technology evolves, the evidence strategy must expand accordingly.
Layer 4: Quality and Risk Management Systems
Quality and risk management systems provide lifecycle governance for innovation-led products. ISO 13485:2016 and ISO 14971:2019 frameworks connect design changes, clinical insights, manufacturing updates, and post-market signals within a single control structure. Therefore, regulatory affairs and design teams should maintain continuous alignment between risk controls, performance data and design outputs across iterations. CAPA, vigilance, and change management processes should guide ongoing product evolution and lifecycle decision-making.
Layer 5: Manufacturing and Scale-Up Readiness
Manufacturing systems for novel technologies must be built alongside product design and EU regulatory expectations. Manufacturing processes should support reliable validation, clear supplier traceability, controlled configuration changes, and readiness for regulatory review and compliance under MDR and IVDR. As production scales, manufacturing plans should maintain design intent and regulatory consistency while accommodating higher volumes and broader market reach.
Layer 6: Notified Body Interaction and Lifecycle Oversight
EU MedTech innovators must engage Notified Bodies throughout product development, approval, and post-market use. Design, manufacturing, and regulatory teams should maintain ongoing technical discussions that address design decisions, evolving evidence, risk management, and lifecycle alignment. As novel products enter routine clinical use and post-market data accumulates, engagement with Notified Bodies should continue in a coordinated manner to capture real-world performance, emerging risks and ongoing lifecycle changes.
Together, these layers establish the execution environment required for advanced EU MedTech innovation to progress seamlessly from development through scale.
Regulation-Enabled Innovation Wins in Europe and Beyond
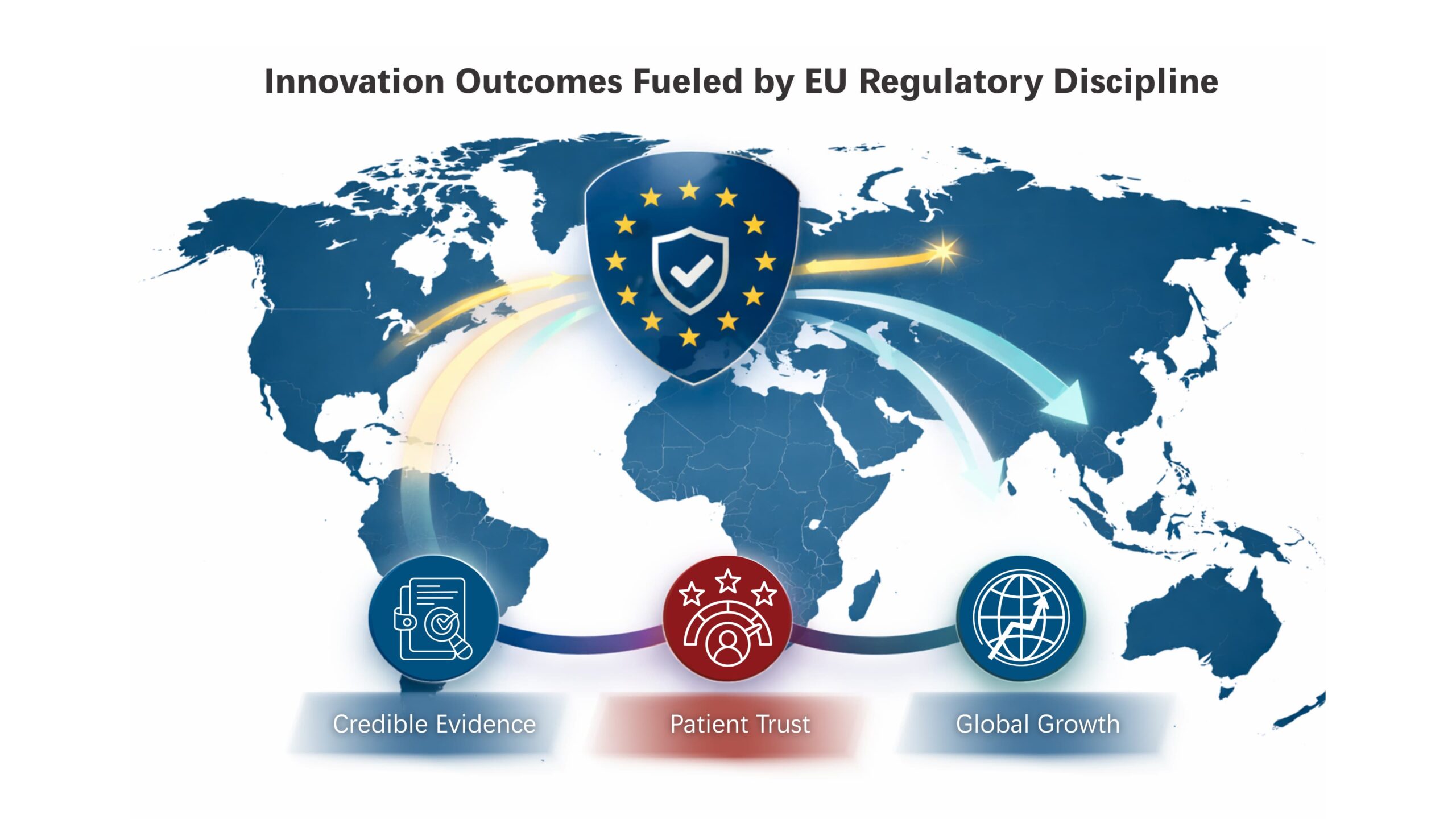
The strength of the EU MedTech ecosystem lies in its ability to align MedTech research and technological advancement with rigorous expectations for safety, clinical performance and quality. This balance enables innovation with lasting clinical impact and global relevance.
Market Credibility Through Evidence-Centric Innovation
In Europe, market credibility is closely linked to the depth and quality of clinical and real-world evidence. EU medical device regulations place sustained emphasis on clinical evaluation, performance monitoring and lifecycle evidence generation, creating products that are defensible not only at approval but throughout market use. This evidence-centric model strengthens credibility with clinicians, payers and regulators globally.
Patient Trust and Safety as Drivers of Long-Term Adoption
Stringent post-market surveillance and vigilance requirements in the EU reinforce patient safety and clinical confidence over time. Continuous monitoring, feedback loops, and structured risk management support responsible product evolution, contributing to sustained patient trust and long-term adoption.
Competitive Advantage in Global Markets
Alignment with EU regulatory expectations positions MedTech products strongly for international expansion. MDR, IVDR, as well as AI and digital health regulations in the EU establish a high bar for evidence quality, lifecycle governance and manufacturing control. Products developed to meet these requirements are often better prepared for regulatory engagement across other global markets, supporting efficient market entry and long-term scalability.
EU MedTech Innovation Requires Specialized Regulatory Integration
Advanced EU MedTech innovation operates within regulatory environments that extend beyond baseline compliance. Furthermore, novel technologies introduce ambiguity across classification, clinical evidence expectations, post-market obligations, and lifecycle oversight. Managing this complexity requires a specific QARA strategy for Europe that operates in parallel with product design, manufacturing, and scale-up.
Generic compliance providers typically struggle with three critical areas that define novel EU MedTech innovation:
- Interpreting ambiguous regulatory guidance for unprecedented technologies.
- Building cross-functional alignment amid uncertain and evolving notified body requirements.
- Developing regulatory strategies that anticipate future policy shifts rather than simply responding to current requirements.
The Syrma Johari MedTech Difference: Navigating Uncharted Regulatory Territory for High-Novelty Devices
End-to-End Execution Across the Innovation Stack
Breakthrough EU MedTech innovation requires regulatory and quality considerations well before formal submission. Syrma Johari MedTech supports execution across development, scale-up, and post-market phases, helping teams align design, evidence, and manufacturing choices with EU medical device regulations. We operate as an integrated MedTech CDMO across the full product value chain: from user requirement definition and design & engineering to verification, regulatory submission support, large volume manufacturing and lifecycle sustenance. Cross-functional teams across design, QARA, and production work within a unified execution framework, reducing handoff risk and ensuring that regulatory, technical, and manufacturing decisions remain in sync throughout development and commercialization.
Core Competency in Handling Regulatory Ambiguity
If existing guidance doesn’t clearly address your technology, we construct defensible regulatory positions grounded in first principles, analogous precedents and proactive engagement with authorities. We have successfully guided novel device classifications, built acceptance for alternative clinical evidence frameworks, and established new precedent cases with notified bodies for emerging technologies.
Multi-Layered EU Compliance Expertise
Innovation-led products often trigger intersecting requirements across MDR, IVDR, EU AI Act, digital health regulations and device-specific legislation. Our team includes specialists who understand the interaction of these regulations in real-world product development. Therefore, we architect regulatory compliance strategies that address the entire regulatory stack, identifying interdependencies and optimizing your pathway across all applicable frameworks.
Long-Horizon Regulatory Planning
Truly innovative devices require a regulatory strategy that extends beyond initial market entry. We build lifecycle roadmaps that account for anticipated guideline updates and future market expansion. For example, developing a clinical trial design supporting both EU approval and future US submission, or a technical documentation structure that accommodates iterative design updates.
European Market Experience and Regional Presence
Syrma Johari MedTech supports multiple EU-focused MedTech engagements with delivery models aligned to MDR and IVDR compliance, including CE technical documentation, design controls, audit preparedness, and notified body interaction support. We maintain operational and business presence in Europe, including Germany and Serbia, alongside our US and India centers. This enables closer collaboration with EU sponsors and faster regulatory coordination. Our global footprint also supports region-aware execution across design, quality assurance and regulatory affairs, and manufacturing initiatives.
Converting Regulatory Uncertainty into a Structured QARA Strategy for Europe
For high-novelty devices, the primary regulatory risk is misalignment across decisions made at different stages of development. Small early assumptions in classification, evidence scope, software architecture, or manufacturing controls can compound into major delays later. Structured regulatory execution reduces this compounding risk and converts uncertainty into a managed pathway.
Cutting-edge EU MedTech innovation demands regulatory execution embedded across the entire product lifecycle, from the first design decision to post-market evolution. Innovators who recognize this early move faster through regulatory complexity and scale with fewer setbacks. Syrma Johari MedTech helps companies build and integrate this regulatory execution capability across the product lifecycle.